



Офев
МНН:
Код ATХ:
L01XE31
Регистрационный номер:
UA/16115/01/01
Срок действия регистрационного удостоверения:
04.07.2017 до 04.07.2022
Название на английском:
OFEV®
- Лікарська форма
- Фармакотерапевтична група
- Фармакологічні властивості
- Діти
- Показання
- Протипоказання
- Взаємодія з іншими лікарськими засобами та інші види взаємодій
- Особливості застосування
- Застосування у період вагітності або годування груддю
- Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
- Спосіб застосування та дози
- Діти
діюча речовина: нінтеданіб;
1 капсула містить 100 мг або 150 мг нінтеданібу (у вигляді езилату);
допоміжні речовини: тригліцериди середньоланцюгові, твердий жир, лецитин (соєвий)
(Е 322);
оболонка капсули: желатин, гліцерин 85%, титану діоксид (Е 171), заліза оксид червоний (Е 172), заліза оксид жовтий (Е 172);
чорнило чорного кольору для маркування капсул:шелак, етанол, пропіленгліколь (Е 1520), заліза оксид чорний (E 172).
Лікарська форма
Капсули м’які.Основні фізико-хімічні властивості:
ОФЕВ, капсули м’які по 100 мг
Продовгуваті непрозорі м’які желатинові капсули персикового кольору, з одного боку чорним чорнилом нанесений логотип компанії «Boehringer Ingelheim» і маркування «100».
Капсули містять в´язку суспензію яскраво-жовтого кольору.
ОФЕВ, капсули м’які по 150 мг
Продовгуваті непрозорі м’які желатинові капсули коричневого кольору, з одного боку чорним чорнилом нанесений логотип компанії «Boehringer Ingelheim» і маркування «150».
Капсули містять в´язку суспензію яскраво-жовтого кольору.
Фармакотерапевтична група
Антинеопластичні засоби. Інгібітори протеїнкінази.Код ATХ L01X E31.
Фармакологічні властивості
Фармакодинаміка.
Механізм дії
Нінтеданіб є низькомолекулярним інгібітором тирозинкінази, який блокує рецептори, у тому числі рецептор фактора росту тромбоцитів (PDGFR) α та β, рецептор фактора росту фібробластів (FGFR) 1-3 і VEGFR 1-3. Нінтеданіб конкурентно взаємодіє з аденозинтрифосфат (АТФ)-зв’язуючою ділянкою цих рецепторів і блокує внутрішньоклітинну передачу сигналів. Крім того, нінтеданіб інгібує кінази Flt-3 (Fms-подібна білкова тирозинкіназа), Lck (лімфоцит-специфічна білкова тирозинкіназа), Lyn (білкова тирозинкіназа lyn) та Src (протоонкогенна білкова тирозинкіназа src).
Нінтеданіб пригнічує активацію сигнальних каскадів FGFR і PDGFR, яка є особливо важлива для проліферації, міграції та диференціації легеневих фібробластів/міофібробластів, характерних клітин в патогенезі ідіопатичного легеневого фіброзу. Потенційний вплив нінтеданібу на інгібування VEGFR та антиангіогенна дія нінтеданібу в патогенезі ідіопатичного легеневого фіброзу (ІЛФ) нині повністю не з’ясована. У доклінічних моделях захворювання на легеневий фіброз нінтеданіб виявляв протифіброзну та протизапальну дію. Нінтеданіб інгібує проліферацію, міграцію та трансформацію фібробластів в міофібробласти у легенях хворих на ідіопатичний легеневий фіброз.
Клінічна ефективність та безпека
Клінічна ефективність нінтеданібу вивчалася у пацієнтів з ІЛФ в рамках двох рандомізованих подвійних сліпих плацебо-контрольованих досліджень фази ІІІ з однаковим дизайном (INPULSIS-1 (1199.32) та INPULSIS-2 (1199,34)). Пацієнти з вихідним розрахунковим значенням ФЖЄЛ
Первинною кінцевою точкою був річний показник скорочення форсованої життєвої ємності легенів (ФЖЄЛ). Ключовими вторинними кінцевими точками були зміна загального балу за анкетою лікарні Святого Георгія для оцінки дихальної функції (SGRQ) на 52-му тижні відносно вихідних значень, а також час до першого загострення ІЛФ.
Річний показник скорочення ФЖЄЛ
Річний показник скорочення ФЖЄЛ (у мл) продемонстрував суттєве зниження у пацієнтів, які отримували нінтеданіб, порівняно з тими, хто отримував плацебо. Лікувальний ефект був подібним в обох дослідженнях (див. табл. 1).
Таблиця 1. Річний показник скорочення ФЖЄЛ у дослідженнях INPULSIS-1, INPULSIS-2 та зведені дані в рамках популяції пацієнтів, що пройшли лікування [[:table:]] [[:table:]]Стабільність ефекту нінтеданібу з точки зору зниження річного показника скорочення ФЖЄЛ була підтверджена в усіх аналізах чутливості за попередньо передбаченими змінними. Первинний аналіз дає можливість припускати, що у пацієнтів, дані яких відсутні, скорочення ФЖЄЛ після останнього зафіксованого значення є аналогічним показникам скорочення, зафіксованим у інших пацієнтів тієї ж групи лікування. В аналізі чутливості, який дав змогу припустити, що у пацієнтів, дані яких відсутні на 52-му тижні, скорочення ФЖЄЛ після останнього зафіксованого значення є аналогічним показникам скорочення, зафіксованим у всіх пацієнтів, які приймали плацебо, скоригована різниця в річному показнику скорочення ФЖЄЛ між нінтеданібом та плацебо склала 113,9 мл/рік (95% ДІ 69,2, 158,5) у дослідженні INPULSIS-1 та 83,3 мл/рік (95% ДІ 37,6, 129,0) у дослідженні INPULSIS-2.
Крім того, аналогічні ефекти спостерігалися щодо інших кінцевих точок для легеневої функції, зокрема зміни ФЖЄЛ на 52-му тижні відносно вихідних значень та аналізів даних пацієнтів, що відповіли на лікування, забезпечуючи тим самим додаткове обґрунтування ефекту нінтеданібу з точки зору уповільнення прогресування захворювання. На рис. 1 продемонстровано процес зміни в часі відносно вихідного значення в обох групах лікування, що ґрунтується на узагальненому аналізі даних, отриманих в ході досліджень INPULSIS-1 та INPULSIS-2.
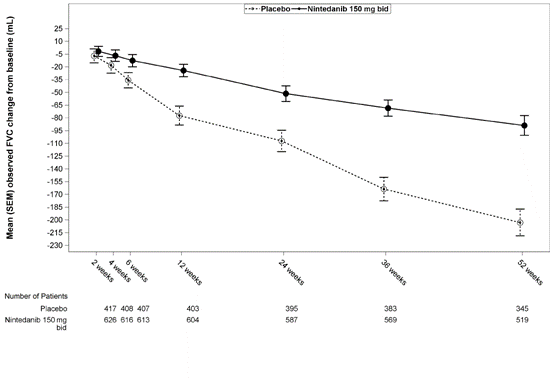
Рис. 1. Середня (SEM) зафіксована зміна ФЖЄЛ в часі відносно вихідного значення (мл), зведені дані досліджень INPULSIS-1 та INPULSIS-2
Аналіз даних пацієнтів, що відповіли на лікування з точки зору показника ФЖЄЛ
В обох дослідженнях INPULSIS відсоток пацієнтів, що відповіли на лікування з точки зору показника ФЖЄЛ, до категорії яких були включені пацієнти, у яких абсолютне розрахункове скорочення ФЖЄЛ у % не перевищило 5% (граничне значення, що вказує на підвищення ризику смертності при ІЛФ), був значно вищим в групі нінтеданібу, ніж у групі плацебо. Аналогічні результати спостерігалися при аналізі із використанням традиційного граничного значення на рівні 10% (див. табл. 2).
Таблиця 2.
Відсоток пацієнтів, що відповіли на лікування з точки зору показника ФЖЄЛ на 52-му тижні у дослідженнях INPULSIS-1, INPULSIS-2 та зведені дані в рамках популяції пацієнтів, що пройшли лікування
[[:table:]]1Пацієнти, що відповіли на лікування – це хворі з абсолютним скороченням ФЖЄЛ не більше ніж на 5 або 10% від розрахункового значення ФЖЄЛ у %, залежно від граничного значення, з оцінкою ФЖЄЛ на 52-му тижні.
2На основі логістичної регресії.
Час до прогресування захворювання (абсолютне скорочення розрахункового значення ФЖЄЛ у % на ≥ 10% або смерть)
В обох дослідженнях INPULSIS було продемонстровано клінічно значуще зниження ризику прогресування захворювання у пацієнтів, що отримували лікування нінтеданібом, у порівнянні із плацебо. У зведеному аналізі показник співвідношення ризиків склав 0,60, вказуючи на 40% зниження ризику прогресування захворювання у пацієнтів, що отримували лікування нінтеданібом, у порівнянні із плацебо.
Таблиця 3. Відсоток пацієнтів з абсолютним скороченням розрахункового значення ФЖЄЛ у % на ≥ 10% або смертю (явища) за період 52 тижні та час до прогресування захворювання у дослідженнях INPULSIS-1, INPULSIS-2 і зведені дані в рамках популяції пацієнтів, що пройшли лікування [[:table:]] [[:table:]]Зміна загального балу SGRQ на 52-му тижні відносно вихідних значень
Загальний бал SGRQ, відповідно до якого вимірюється пов’язана зі здоров’ям якість життя (HRQoL), був проаналізований на 52-му тижні. У дослідженні INPULSIS-2 пацієнти, які отримували плацебо, продемонстрували суттєвіше збільшення загального балу SGRQ відносно вихідних значень, порівняно із пацієнтами, які приймали нінтеданіб в дозі 150 мг двічі на добу. Погіршення показника HRQoL було меншим у групі нінтеданібу; різниця між двома групами лікування була статистично значущою (-2,69; 95% ДІ: -4,95, -0,43; p=0.0197).
У дослідженні INPULSIS-1 збільшення загального балу SGRQ на 52-му тижні відносно вихідних значень було порівнянним у групі нінтеданібу та групі плацебо (різниця між групами лікування: -0,05; 95% ДІ: -2,50, 2,40; p=0,9657). У зведеному аналізі досліджень INPULSIS розрахункова середня зміна загального балу SGRQ на 52-му тижні відносно вихідних значень була меншою в групі нінтеданібу (3,53), ніж в групі плацебо (4,96), з різницею між групами лікування на рівні -1,43 (95% ДІ: -3,09, 0,23; p=0,0923). Загалом, вплив нінтеданібу на пов’язану зі здоров’ям якість життя, що вимірюється на основі загального балу SGRQ, є незначним та демонструє менше погіршення порівняно із плацебо.
Час до першого загострення ІЛФ
У дослідженні INPULSIS-2 ризик першого загострення ІЛФ за 52-тижневий період був суттєво нижчим у пацієнтів, які отримували нінтеданіб, порівняно з тими, хто отримував плацебо, а у дослідженні INPULSIS-1 між групами лікування різниці не було. У зведеному аналізі досліджень INPULSIS у пацієнтів, які отримували нінтеданіб, спостерігався чисельно менший ризик першого загострення, порівняно з тими, хто приймав плацебо (див. табл. 4).
Таблиця 4. Відсоток пацієнтів із загостреннями ІЛФ (явища) за період 52 тижні та час до першого загострення на основі даних, повідомлених дослідником, у дослідженнях INPULSIS-1, INPULSIS-2 і зведені дані в рамках популяції пацієнтів, що пройшли лікування [[:table:]] [[:table:]]Усі небажані явища, виражені у формі загострень ІЛФ, про які повідомляв дослідник, проходили експертну оцінку в експертному комітеті, що працює у «сліпому» режимі. Сенситивний аналіз часу до першого підтвердженого або підозрюваного випадку загострення ІЛФ, що пройшов експертну оцінку, за попередньо передбаченими змінними проводився на зведених даних. Відсоток пацієнтів, які мали принаймні 1 випадок загострення, що пройшов експертну оцінку, протягом 52 тижнів, був нижчим у групі нінтеданібу (1,9% пацієнтів), ніж у групі плацебо (5,7% пацієнтів). В аналізі часу до настання оціненого випадку загострення із використанням зведених даних було виведено значення співвідношення ризиків (HR) на рівні 0,32 (95% ДІ 0,16, 0,65; p=0,0010). Це вказує на те, що ризик виникнення першого загострення ІЛФ був статистично значуще нижчим у групі нінтеданібу, ніж у групі плацебо в будь-який момент часу.
Аналіз виживаності
У зведеному аналізі даних виживаності за попередньо визначеними змінними в рамках досліджень INPULSIS загальна смертність за 52-тижневий період була нижчою у групі нінтеданібу (5,5%), ніж в групі плацебо (7,8%). В аналізі часу до смерті було визначено значення HR – 0,70 (95% ДІ 0,43, 1,12; p=0,1399). Результати усіх кінцевих точок за параметром «виживаність» (таких як смертність в процесі лікуванні та смертність від респіраторних явищ) продемонстрували переконливу чисельну різницю на користь нінтеданібу.
Таблиця 5. Смертність з усіх причин (явища) за період 52 тижні у дослідженнях INPULSIS-1, INPULSIS-2 і зведені дані в рамках популяції пацієнтів, що пройшли лікування [[:table:]] [[:table:]]Додаткові дані на підтримку, отримані в рамках дослідження II фази (1199.30) препарату ОФЕВ в дозі 150 мг двічі на добу
Додаткові дані, що свідчать на користь ефективності препарату, були отримані в ході рандомізованого, подвійного сліпого, плацебо-контрольованого дослідження фази ІІ з підбором доз за участю групи пацієнтів, які приймали нінтеданіб в дозі 150 мг двічі на добу.
Показник скорочення ФЖЄЛ протягом 52 тижнів, що був первинною кінцевою точкою дослідження, був нижчим в групі нінтеданібу (-0,060 л/рік, N=84), ніж в групі плацебо
(-0,190 л/рік, N=83). Розрахункова різниця між групами лікування склала 0,131 л/рік (95% ДІ 0,027, 0,235). Різниця між групами лікування досягла номінальної клінічної значущості (p=0,0136).
Розрахункова середня зміна загального балу SGRQ на 52-му тижні відносно вихідних значень склала 5,46 для плацебо, що вказує на погіршення пов’язаної зі здоров’ям якості життя, та -0,66 для нінтеданібу, що вказує на стабільну пов’язану зі здоров’ям якість життя. Розрахункова середня зміна для нінтеданібу порівняно з плацебо склала -6,12 (95% ДІ: -10,57, -1,67; p=0,0071).
Кількість пацієнтів із загостреннями ІЛФ за 52-тижневий період була нижчою в групі нінтеданібу (2,3%, N=86), ніж в групі плацебо (13,8%, N=87). Розрахункове значення співвідношення ризиків нінтеданібу проти плацебо склало 0,16 (95% ДІ 0,04, 0,71; p=0,0054).
Інтервал QT
В рамках спеціального дослідження за участю пацієнтів з нирково-клітинним раком були зроблені вимірювання інтервалу QT/комплексу QT; в результаті цих вимірювань встановлено, що разова пероральна доза 200 мг нінтеданібу, а також багаторазові пероральні дози 200 мг нінтеданібу із прийомом в режимі двічі на добу протягом 15 днів не подовжувати інтервал QT з коригуванням Фрідеріція.
Діти
ОФЕВ не досліджували у педіатричній практиці при ІЛФ.
Фармакокінетика.
Абсорбція
Максимальна концентрація нінтеданібу в плазмі крові досягається приблизно через 2–4 години після перорального прийому препарату у формі м’яких желатинових капсул під час їди (діапазон 0,5–8 годин). Абсолютна біодоступність дози 100 мг складає у здорових добровольців 4,69% (90% ДІ: 3,615–6,078). Абсорбція та біодоступність зменшуються внаслідок дії транспортера та суттєвого пресистемного метаболізму. Встановлено, що експозиція нінтеданібу збільшується пропорційно дозі (у діапазонах доз 50–450 мг один раз на добу та 150–300 мг двічі на добу). Стійкі концентрації в плазмі крові досягаються, як максимум, впродовж одного тижня після початку прийому.
Експозиція нінтеданібу збільшується після їди приблизно на 20% у порівнянні з прийомом препарату натще (ДІ: 95,3–152,5%), а всмоктування сповільнюється (медіана часу досягнення максимальної концентрації в плазмі крові натще (tmax) – 2,00 години; після їди – 3,98 години).
Розподіл
Розподіл нінтеданібу здійснюється шляхом двофазної кінетики. Після внутрішньовенної інфузії під час термінальної фази спостерігається великий об’єм розподілу (Vss): 1050 л, геометричний коефіцієнт варіації (gCV) 45,0%).
Зв’язування нінтеданібу з білками плазми людини in vitro вважається значним, пов’язана фракція складає 97,8%. Основним білком, що бере участь в зв’язуванні, вважається альбумін сироватки крові. Нінтеданіб переважно розподіляється в плазмі, співвідношення кров/плазма складає 0,869.
Біотрансформація
Основною реакцією, що бере участь в метаболізмі нінтеданібу, є гідролітичне розщеплювання за допомогою естераз, що призводить до утворення вільного кислого метаболіту нінтеданібу (BIBF 1202). Надалі BIBF 1202 глюкуронізується ферментами уридін-5’-дифосфат-глюкуронозилтрасферази (UGT), а саме UGT 1A1, UGT 1A7, UGT 1A8 та UGT 1A10, з утворенням глюкуроніду BIBF 1202.
Біотрансформація нінтеданібу за участю ізоферментів CYP відбувається лише незначною мірою; основну роль у цьому процесі відіграє ізофермент CYP 3A4. У дослідженні ADME у людини основний метаболіт, що утворюється за участю ізоферментів CYP, виявити в плазмі не вдалося. За даними дослідження in vitro CYP-залежний метаболізм складає приблизно 5%, тоді як розщеплювання, здійснюване естеразами, складає 25%. Нінтеданіб, BIBF 1202 та глюкуронід BIBF 1202 не пригнічували і не стимулювали ізоферменти CYP і в доклінічних дослідженнях. Тому не слід очікувати лікарських взаємодій між нінтеданібом і субстратами CYP, інгібіторами CYP або індукторами CYP.
Виведення
Загальний кліренс плазми після внутрішньовенної інфузії є високим (1390 мл/хв, gCV – 28,8%). Виведення із сечею незміненої активної речовини впродовж 48 годин після прийому нінтеданібу внутрішньо складає приблизно 0,05% від величини дози (gCV – 31,5%), а після внутрішньовенного введення – приблизно 1,4% (gCV – 24,2%); нирковий кліренс складає 20 мл/хв (gCV – 32,6%). Після прийому внутрішньо [14C]-нінтеданібу радіоактивний матеріал виводився переважно з жовчю і виявлявся в калі (93,4% дози, gCV – 2,61%). Частка ниркової екскреції в загальному кліренсі є низькою (0,649% дози (gCV – 26,3%)). Виведення вважається повним (більше 90%) через 4 дні після прийому. Період напіввиведення нінтеданібу в термінальній стадії складає від 10 до 15 годин (gCV приблизно 50%).
Лінійність/нелінійність
Можна припустити, що фармакокінетика (ФК) нінтеданібу лінійна відносно часу (тобто дані щодо застосування разової дози можуть бути екстрапольовані на дані щодо багаторазового використання). Значення Cmax в результаті накопичення препарату після багаторазового застосування перевищує показник Cmax разової дози в 1,04 раза, а значення AUCτ – в 1,38 раза. Мінімальні остаточні концентрації нінтеданібу залишаються стабільними протягом більше одного року.
Транспортування
Нінтеданіб є субстратом для глікопротеїну P (P-gp). Див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій» для отримання інформації стосовно можливої взаємодії нінтеданібу із цим транспортером. Показано, що нінтеданіб in vitro не є субстратом або інгібітором OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 або MRP-2. Нінтеданіб також не є субстратом BCRP. In vitro було встановлено, що нінтеданіб має слабку інгібувальну активність відносно OCT-1, BCRP і P-gp, що, як вважається, має невелику клінічну значущість. Такий же висновок зроблений стосовно нінтеданібу як субстрату OCT-1.
Фармакокінетика у особливих груп пацієнтів
Фармакокінетичні властивості нінтеданібу були порівнянні у здорових добровольців, пацієнтів з ІЛФ та онкологічними захворюваннями. З огляду на результати популяційного фармакокінетичного аналізу у пацієнтів з ІЛФ та недрібноклітинним раком легенів (НДРЛ) (N=1 191) і описових досліджень, на дію нінтеданібу не впливали такі чинники, як стать (з поправкою на масу тіла), порушення функції нирок легкого і середнього ступеня тяжкості (розраховане на основі кліренсу креатиніну), вживання алкоголю або генотип Р-глікопротеїну. Популяційний фармакокінетичний аналіз виявив помірний вплив статі, маси тіла і расової приналежності на дію нінтеданібу, як описано нижче. У зв’язку з тим, що спостерігалася висока міжіндивідуальна варіабельність експозиції, ці несуттєві впливи не вважалися клінічно значущими (див. розділ «Особливості застосування»).
Вік
Експозиція нінтеданібу лінійно збільшується з віком. У 45-річних пацієнтів значення AUCτ,ss було нижче на 16%, а у 76-річних пацієнтів вище на 13% в порівнянні з пацієнтами, медіана віку яких складала 62 роки. Діапазон віку, що оцінювався в ході аналізу, складав 29-85 років; вік більше 75 років відзначався приблизно у 5% популяції пацієнтів. На основі моделі популяційного фармакокінетичного аналізу встановлено, що у пацієнтів віком від 75 років спостерігалося збільшення експозиції нінтеданібу приблизно на 20–25%, порівняно із пацієнтами віком до 65 років.
Аналогічні дослідження у дітей не проводилися.
Маса тіла
Спостерігається зворотна кореляція між масою тіла і експозицією нінтеданібу. У пацієнтів з масою тіла 50 кг (5-й процентиль) величина AUCτ,ss збільшувалася на 25%, а у пацієнтів з масою тіла 100 кг (95-й процентиль) зменшувалася на 19% у порівнянні з пацієнтами, медіана маси тіла яких складала 71,5 кг.
Раса
Середня геометрична експозиція нінтеданібу на 33% вища у пацієнтів-китайців, тайванців та індусів, а у пацієнтів-корейців – на 22% нижча, ніж у пацієнтів європеоїдної раси (з поправкою на масу тіла). Дані стосовно пацієнтів негроїдної раси є дуже обмеженими; діапазон цих даних схожий на такий у пацієнтів європеоїдної раси.
Порушення функції печінки
Фармакокінетичні показники нінтеданібу були отримані у пацієнтів з порушеннями показників функції печінки (підвищення АСТ, АЛТ і концентрації білірубіну). У пацієнтів з початковим підвищенням рівнів АСТ та АЛТ (до 10 разів в порівнянні з верхньою межею норми) та концентрації білірубіну (до 1,5 раза в порівнянні з верхньою межею норми) спостерігалася тенденція до збільшення експозиції нінтеданібу (у порівнянні з пацієнтами з нормальними значеннями АСТ, АЛТ і білірубіну). Обмежені дані про пацієнтів з підвищеною активністю АЛТ або АСТ, що перевищувала верхню межу норми більше ніж в 10 разів, і підвищенням концентрації білірубіну, що перевищувало верхню межу норми більше ніж в 1,5 раза, не дали змогу зробити остаточні висновки.
Одночасна терапія з пірфенідоном
У невеликому дослідженні з паралельними групами за участю японських пацієнтів з ІЛФ (13 пацієнтів отримували нінтеданіб одночасно зі стандартними дозами пірфенідону в рамках тривалого лікування; 11 пацієнтів отримували лише нінтеданіб) експозиція нінтеданібу зменшилася до 68,3% на основі показника AUC та до 59,2% на основі показника Cmax в результаті одночасного застосування нінтеданібу з пірфенідоном, порівняно з монотерапією нінтеданібом. Нінтеданіб не впливав на фармакокінетику пірфенідону (див. розділ «Особливості застосування»).
Клінічні характеристики
Показання
ОФЕВ показаний для лікування ідіопатичного легеневого фіброзу (ІЛФ) у дорослих.
Протипоказання
Підвищена чутливість до нінтеданібу, арахісу чи сої або до будь-якої із допоміжних речовин препарату.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
P-глікопротеїн (P-gp)
Нінтеданіб є субстратом P-gp (див. розділ «Фармакологічні властивості. Фармакокінетика»). У спеціальному дослідженні взаємодії препаратів встановлено, що спільне застосування з активним інгібітором P-gp кетоконазолом збільшує експозицію нінтеданібу, судячи з величини AUC, в 1,61 разу, а за показником Cmax в 1,83 раза. Спеціальне дослідження взаємодії препаратів продемонструвало, що одночасне застосування рифампіцину (активного індуктора P-gp) призводить до зменшення експозиції нінтеданібу, за показником AUC на 50,3%, а за показником Cmax на 60,3% (в порівнянні із застосуванням одного нінтеданібу). Активні інгібітори P-gp (наприклад кетоконазол, еритроміцин або циклоспорин) у разі спільного застосування з препаратом ОФЕВ можуть збільшувати експозицію нінтеданібу. У таких пацієнтів переносимість нінтеданібу потрібно ретельно відслідковувати. При виникненні побічних реакцій може бути потрібне призупинення терапії, зниження дози або відміна лікування препаратом ОФЕВ (див. розділ «Спосіб застосування та дози»).
Активні індуктори P-gp (наприклад рифампіцин, карбамазепін, фенітоїн і препарати звіробою звичайного) можуть зменшувати експозицію нінтеданібу. Рекомендується підбір альтернативної супутньої терапії з відсутністю або мінімальною індукуючою дією на систему P-gp.
Ізофермент цитохрому (CYP)
Ізоферменти CYP беруть лише незначну участь у біотрансформації нінтеданібу. У доклінічних дослідженнях нінтеданіб та його метаболіти (BIBF 1202 – вільний кислий метаболіт нінтеданібу і його глюкуронід BIBF 1202) не інгібували і не індукували ізоферменти CYP (див. розділ «Фармакологічні властивості. Фармакокінетика»). Тому вірогідність лікарських взаємодій з нінтеданібом, що ґрунтуються на метаболізмі CYP, вважається невеликою.
Одночасне застосування з іншими препаратами
Можливість взаємодій нінтеданібу з гормональними контрацептивними засобами не вивчалася.
Особливості застосування
Порушення з боку шлунково-кишкового тракту
Діарея
У дослідженнях INPULSIS (див. розділ «Фармакологічні властивості. Фармакодинаміка») діарея була найбільш частим побічним явищем з боку шлунково-кишкового тракту і відзначалася у 62,4% пацієнтів, які отримували препарат ОФЕВ, в порівнянні з 18,4% пацієнтів, які отримували плацебо (див. розділ «Побічні реакції»). У більшості пацієнтів ці небажані явища були легкого і помірного ступеня тяжкості і відзначалися упродовж перших 3 місяців лікування. Діарея стала причиною зниження дози нінтеданібу у 10,7% пацієнтів та припинення терапії нінтеданібом у 4,4% пацієнтів.
Лікування діареї (адекватна гідратація та антидіарейні лікарські засоби, наприклад, лоперамід) слід проводити при появі перших ознак. У разі розвитку діареї може бути потрібне переривання лікування. Лікування препаратом ОФЕВ можна відновити у зниженій дозі (100 мг двічі на добу) або повній дозі (150 мг двічі на добу). У разі продовження тяжкої діареї, незважаючи на симптоматичне лікування, терапію препаратом ОФЕВ слід відмінити.
Нудота та блювання
Нудота і блювання були небажаними явищами з боку шлунково-кишкового тракту, про які часто повідомлялося (див. розділ «Побічні реакції»). У більшості пацієнтів відзначалася нудота та блювання легкого або помірного ступеня тяжкості. Нудота стала причиною припинення лікування нінтеданібом у 2,0% пацієнтів. Блювання стало причиною припинення лікування у 0,8% пацієнтів.
Якщо симптоми не зникають, незважаючи на належну симптоматичну терапію (включаючи застосування протиблювотних засобів), може бути потрібне зменшення дози препарату або призупинка лікування. Лікування можна відновити у зниженій дозі (100 мг двічі на добу) або повній дозі (150 мг двічі на добу). Якщо тяжкі симптоми не зникають, терапію препаратом ОФЕВ слід відмінити.
Порушення функції печінки
У пацієнтів з порушеннями функції печінки помірного (клас B за шкалою Чайлда-П’ю) і тяжкого (клас C за шкалою Чайлда-П’ю) ступеня безпечність та ефективність препарату ОФЕВ не вивчалися. Тому лікування таких пацієнтів препаратом ОФЕВ не рекомендується (див. розділи «Спосіб застосування та дози»). З огляду на посилену дію лікарського засобу ризик небажаних реакцій може збільшуватися у пацієнтів із порушеннями функції печінки легкого ступеню (клас А за шкалою Чайлда-П’ю). Для пацієнтів із порушеннями функції печінки легкого ступеню (клас А за шкалою Чайлда-П’ю) необхідно призначати лікування зменшеною дозою ОФЕВ (див. розділи «Спосіб застосування та дози» та «Фармакологічні властивості. Фармакокінетика»)
Застосування нінтеданібу було пов’язане із підвищенням рівня печінкових ферментів (АЛТ, АСТ, лужної фосфатази (ЛФ), гамма-глутамілтрансферази (ГГТ); при цьому більший ризик підвищення рівня ферментів можливий у жінок. Підвищення рівня трансаміназ було оборотним, цей показник повертався до початкових значень після зменшення дози або відміни препарату. Застосування нінтеданібу також було пов’язане із підвищенням рівня білірубіну та ураженням печінки, спричиненим препаратом. Рекомендується оцінювати активність печінкових трансаміназ і концентрацію білірубіну до початку терапії препаратом ОФЕВ, а потім періодично під час лікування (наприклад при кожному візиті пацієнта) або за клінічними показаннями. У разі підвищення рівня трансаміназ (АСТ або АЛТ) більш ніж в 3 рази вище за верхню межу норми рекомендовано зменшити дозу або перервати терапію препаратом ОФЕВ і проводити пильне спостереження за пацієнтом. Як тільки показники трансаміназ повернуться до початкового рівня, лікування препаратом ОФЕВ можна відновити в повній дозі (150 мг двічі на добу) або в зниженій дозі (100 мг двічі на добу), яка згодом може бути підвищена до повної дози (див розділ «Спосіб застосування та дози»). Якщо підвищення будь-яких показників функції печінки пов’язане з клінічними симптомами ураження печінки, наприклад, жовтяницею, лікування препаратом ОФЕВ слід остаточно припинити. Потрібно досліджувати альтернативні причини підвищення рівнів печінкових ферментів.
Кровотечі
Пригнічення рецептора судинного ендотеліального фактора росту (VEGFR) може бути пов’язано із підвищеним ризиком кровотечі. У дослідженнях INPULSIS лікарського засобу ОФЕВ відсоток пацієнтів, у яких були відзначені небажані явища у вигляді кровотеч, був дещо вищим у групі прийому ОФЕВ (10,3%), ніж у групі плацебо (7,8%). Легкі носові кровотечі були найбільш частим небажаним явищем серед випадків кровотечі. Випадки серйозних кровотеч відзначалися рідко і з практично однаковою частотою у двох групах лікування (плацебо: 1,4%; ОФЕВ: 1,3%).
У дослідження INPULSIS не включали пацієнтів з відомим ризиком розвитку кровотеч, у т.ч. пацієнтів зі спадковою схильністю до кровотеч або пацієнтів, які одержували антикоагулянтну терапію у високих дозах. Повідомлялося про випадки кровотеч у післяреєстраційному періоді (такі повідомлення стосувалися як пацієнтів, які одержували антикоагулянтну терапію або інші лікарські засоби, що могли спричинити кровотечу, так і пацієнтів, які не приймали такі засоби). Отже, цій категорії пацієнтів лікування препаратом ОФЕВ можна бути призначати тільки тоді, коли потенційна користь терапії перевищує потенційний ризик.
Артеріальна тромбоемболія
З клінічних досліджень INPULSIS виключали пацієнтів з інфарктом міокарда або інсультом в найближчому анамнезі. Випадки розвитку артеріальної тромбоемболії зустрічалися рідко: у 0,7% пацієнтів, що отримували плацебо, і 2,5% пацієнтів в групі, що отримувала нінтеданіб. Тоді як небажані реакції, що відображають ішемічні захворювання серця, були порівнянні в групах нінтеданібу і плацебо, пацієнтів, у яких розвинувся інфаркт міокарда, виявилося більше в групі нінтеданібу (1,6%) у порівнянні з групою плацебо (0,5%). Необхідно дотримуватися обережності при лікуванні пацієнтів з високим серцево-судинним ризиком, включаючи відоме захворювання коронарних артерій. Слід розглянути можливість перерви в лікуванні пацієнтів, у яких розвинулися симптоми гострої міокардіальної ішемії.
Венозна тромбоемболія
У дослідженнях INPULSIS не спостерігалося підвищеного ризику розвитку венозних тромбоемболічних ускладнень у пацієнтів, які приймали нінтеданіб. Проте у зв’язку з особливостями механізму дії нінтеданібу у пацієнтів можливе відзначатися підвищення ризику розвитку тромбоемболічних явищ.
Перфорації шлунково-кишкового тракту (ШКТ)
У дослідженнях INPULSIS не спостерігалося підвищеного ризику розвитку перфорацій ШКТ у пацієнтів, які приймали нінтеданіб. Проте у зв’язку з особливостями механізму дії нінтеданібу у пацієнтів може підвищуватися ризик розвитку перфорацій ШКТ. Особливу увагу слід приділяти лікуванню пацієнтів, яким раніше проводили абдомінальні хірургічні втручання. У зв’язку з цим ОФЕВ можна застосовувати лише як мінімум через 4 тижні після абдомінальних хірургічних втручань. У разі виникнення перфорації ШКТ терапію препаратом ОФЕВ потрібно припиняти.
Артеріальна гіпертензія
Прийом препарату ОФЕВ може підвищувати артеріальний тиск, тому слід періодично та за клінічними показниками вимірювати артеріальний тиск.
Порушення загоєння ран
У дослідженнях INPULSIS збільшення частоти порушень загоєння ран не спостерігалося. З огляду на механізм дії нінтеданібу, ця речовина може негативно впливати на загоєння ран. Спеціальних досліджень, в яких вивчався б вплив нінтеданібу на загоєння ран, не проводилося. Тому лікування препаратом ОФЕВ має починатися або поновлюватися (якщо здійснювалася перерва у зв’язку з хірургічним втручанням) з урахуванням клінічної думки про адекватність загоєння рани.
Одночасна терапія з пірфенідоном
Одночасне призначення нінтеданібу з пірфенідоном було вивчене у дослідженні з паралельними групами японських пацієнтів з ІЛФ. 24 пацієнти отримували 150 мг нінтеданібу двічі на добу (13 пацієнтів отримували нінтеданіб одночасно зі стандартними дозами пірфенідону в рамках тривалого лікування; 11 пацієнтів отримували лише нінтеданіб). У зв’язку з коротким періодом одночасної дії і малою кількістю пацієнтів, жодні висновки про переваги/ризики комбінованого лікування із пірфенідоном не можуть бути зроблені.
Вплив на інтервал QT
Жодних ознак подовження інтервалу QT при застосуванні нінтеданібу в рамках програми клінічних досліджень не виявлено (див. розділ «Фармакологічні властивості. Фармакодинаміка»). Оскільки відомо, що деякі інші інгібітори тирозинкінази впливають на QT, слід з обережністю призначати нінтеданіб пацієнтам, які знаходяться в групі ризику подовження інтервалу QT.
Алергічні реакції
Відомо, що продукти лікувального харчування з вмістом сої спричиняють алергічні реакції, у тому числі тяжкий анафілактичний шок, в осіб із алергією на сою. Пацієнти з відомою алергією на арахісовий білок знаходяться в групі ризику розвитку тяжких реакцій на препарати із вмістом сої. 1 капсула по 100 мг містить лецитину соєвого 1,2 мг; 1 капсула по 150 мг містить лецитину соєвого – 1,8 мг.
Застосування у період вагітності або годування груддю
Жінки репродуктивного віку
Нінтеданіб може чинити негативний вплив на плід людини. Жінкам репродуктивного віку під час лікування препаратом ОФЕВ слід вживати запобіжних заходів для уникнення вагітності. Їм потрібно порадити використовувати надійні методи контрацепції під час застосування препарату і протягом щонайменше 3 місяців після прийому останньої дози препарату ОФЕВ. Оскільки дія нінтеданібу на метаболізм та ефективність гормональних контрацептивів не вивчалася, методом вибору запобігання вагітності повинен стати бар’єрний метод.
Вагітність
Спеціальних досліджень щодо застосування препарату ОФЕВ під час вагітності у людини не проводилося, проте в доклінічних дослідженнях у тварин встановлена репродуктивна токсичність цієї активної речовини. Оскільки нінтеданіб може мати ембріотоксичну дію у людини, його не слід застосовувати під час вагітності.
Пацієнткам слід негайно повідомити лікаря про розвиток вагітності під час терапії препаратом ОФЕВ.
Якщо під час терапії препаратом ОФЕВ розвивається вагітність, пацієнтку необхідно проінформувати про потенційну небезпеку ембріотоксичної дії препарату. Також слід розглянути питання про припинення лікування препаратом ОФЕВ.
Годування груддю
Відсутні дані про виділення нінтеданібу і його метаболітів в грудне молоко людини. У доклінічних дослідженнях показано, що у тварин в період лактації в грудне молоко проникає невелика кількість нінтеданібу та його метаболітів (≤ 0,5% від величини дози, що застосовувалася). Тому не можна виключити ризик для новонароджених і грудних дітей. Під час лікування препаратом ОФЕВ годування груддю слід припинити.
Фертильність
У доклінічних дослідженнях ознак порушень фертильності у самців виявлено не було. У дослідженнях підгострої та хронічної токсичності, під час яких рівень системної дії препарату був порівнянний з рівнем, що досягається при використанні максимальної рекомендованої дози у людини (150 мг двічі на добу), ознак порушень фертильності у самок тварин виявлено не було.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
ОФЕВ має незначний вплив на здатність керувати транспортним засобом або іншими механізмами. Під час застосування препарату ОФЕВ пацієнтам потрібно рекомендувати дотримуватися обережності при управлінні транспортними засобами або іншими механізмами.
Спосіб застосування та дози
Лікування препаратом ОФЕВ слід розпочинати та проводити під наглядом лікаря, який має досвід діагностики та лікування ІЛФ.
Дози
Рекомендована доза препарату становить 150 мг двічі на добу, приблизно через кожні 12 годин. Доза 100 мг двічі на добу рекомендується лише тим пацієнтам, які погано переносять дозу 150 мг двічі на добу.
Якщо будь-яка доза препарату була пропущена, то слід продовжити прийом препарату в початково рекомендованій дозі за розкладом наступного прийому препарату. Якщо доза була пропущена, пацієнт не повинен приймати додаткову дозу препарату. Максимальна добова доза складає 300 мг.
Коригування дози
При розвитку небажаних реакцій на препарат ОФЕВ (див. розділи «Особливості застосування», «Побічні реакції») на додаток до симптоматичної терапії у разі необхідності рекомендується зниження дози або тимчасове переривання лікування доти, доки небажана реакція не знизиться до рівня, дасть змогу відновити терапію. Лікування препаратом ОФЕВ може бути відновлене в повній дозі (150 мг двічі на добу) або в зниженій дозі (100 мг двічі на добу). Якщо пацієнт не переносить дозу препарату 100 мг двічі на добу, лікування препаратом ОФЕВ слід припинити.
У разі підвищення рівня аспартат-амінотрансферази (АСТ) або аланінамінотрансферази (АЛТ) більш ніж в 3 рази вище за верхню межу норми рекомендовано перервати терапію препаратом ОФЕВ. Як тільки показники повернуться до нормального значення, лікування препаратом ОФЕВ можна відновити в зниженій дозі (100 мг двічі на добу), яка згодом може бути збільшена до повної дози (150 мг двічі на добу) (див. розділи «Особливості застосування», «Побічні реакції»).
Особливі групи пацієнтів
Пацієнти літнього віку ( 65 років)
Не відзначено жодних загальних відмінностей з точки зору безпеки та ефективності застосування препарату літнім пацієнтам. Коригування дози препарату залежно від віку пацієнта не потрібне. Пацієнтам віком від 75 років може бути потрібне зниження дози для мінімізації небажаних явищ (див. розділ «Фармакологічні властивості. Фармакокінетика»).
Порушення функції нирок
Нирками виводиться менше 1% разової дози нінтеданібу (див. розділ «Фармакологічні властивості. Фармакокінетика»). Пацієнтам з порушеннями функції нирок легкого або помірного ступеня тяжкості коригувати початкову дозу не потрібно. Для пацієнтів з тяжкими порушеннями функції нирок (кліренс креатиніну
Порушення функції печінки
Нінтеданіб виводиться переважно з жовчю/калом (> 90%). Дія посилюється у пацієнтів із порушенням функції печінки (клас А та клас В за шкалою Чайлда–П’ю; див. розділ «Фармакологічні властивості. Фармакокінетика»). У пацієнтів з порушеннями функції печінки легкого ступеня тяжкості (клас А за шкалою Чайлда–П’ю) рекомендована доза ОФЕВ становить 100 мг двічі на день приблизно із 12 годинним інтервалом. Для пацієнтів із легким ступенем тяжкості порушення функції печінки (клас А за шкалою Чайлда–П’ю) необхідно передбачити переривання або припинення лікування для контролю за небажаними реакціями. У пацієнтів з порушеннями функції печінки класів B і C за шкалою Чайлда–П’ю безпека та ефективність нінтеданібу не вивчалися. Тому лікування пацієнтів з порушеннями функції печінки помірного (клас B за шкалою Чайлда–П’ю) і тяжкого (клас C за шкалою Чайлда–П’ю) ступеня препаратом ОФЕВ не рекомендується (див. розділ «Фармакологічні властивості. Фармакокінетика»).
Дитячий вік
Безпечність та ефективність застосування препарату ОФЕВ дітям (віком до 18 років) не вивчалися. Дані відсутні.
Спосіб застосування
ОФЕВ призначений для перорального застосування. Капсули слід приймати з їжею, ковтати цілими, запиваючи водою; їх не потрібно розжовувати чи подрібнювати.
Діти
Препарат не застосовують у педіатричній практиці.
Передозування
Симптоми
Зафіксовані випадки передозування у двох пацієнтів, які брали участь в онкологічній програмі, при застосуванні препарату в максимальній дозі 600 мг впродовж восьми днів. Небажані явища, що спостерігалися, були порівнянні з відомим профілем безпеки нінтеданібу: збільшення активності ферментів печінки і порушення з боку ШКТ. Обидва пацієнти повністю відновилися після небажаних явищ. У дослідженнях INPULSIS був зафіксований один випадок ненавмисного підвищення дози пацієнтом до 600 мг на добу упродовж 21 дня. За період неправильного прийому препарату було зафіксовано розвиток небажаного явища (назофарингіт) легкого ступеня, яке пройшло в цьому періоді без фіксування будь-яких інших небажаних реакцій.
Лікування
Специфічного антидоту немає. У разі передозування необхідно відмінити препарат і проводити симптоматичну терапію.
Побічні реакції
Короткий опис профілю безпеки
Нінтеданіб вивчався у клінічних дослідженнях за участю 1 529 пацієнтів з ІЛФ. Представлені дані з безпеки ґрунтуються на двох рандомізованих подвійних сліпих плацебо-контрольованих дослідженнях фази III за участю 1 061 пацієнта, у ході яких порівнювалося лікування нінтеданібом в дозі 150 мг двічі на добу та плацебо упродовж 52 тижнів (INPULSIS-1 та INPULSIS-2).
Найчастішими ПР, пов’язаними із застосуванням нінтеданібу, були діарея, нудота і блювання, біль в ділянці живота, зниження апетиту, зниження маси тіла і підвищення рівня ферментів печінки.
Для одержання інформації стосовно лікування деяких небажаних явищ див. також розділ «Особливості застосування».
Перелік побічних реакцій (ПР)
У таблиці 6 надані побічні реакції відповідно до класів систем органів за MedDRA та за частотою виникнення.
В таблиці 6 підсумована частота побічних реакцій, про які повідомлялося у групі пацієнтів (638), що приймали нінтеданіб в рамках двох плацебо-контрольованих клінічних досліджень фази III тривалістю 52 тижні або у постмаркетингових дослідженнях.
Класифікація частоти виникнення побічних реакцій: дуже часті (>1/10); часті (>1/100 до >1/1 000 до > 1/10 000 до
Таблиця 6.
Резюме побічних реакцій на лікарський засіб за категоріями частоти [[:table:]]Опис окремих побічних реакцій
Діарея
Діарея відзначалася у 62,4% пацієнтів, які отримували нінтеданіб. У 3,3% пацієнтів, що приймали нінтеданіб, була зафіксована тяжка діарея. У більше ніж двох третин пацієнтів діарея відзначалася упродовж перших 3 місяців лікування. Діарея стала причиною остаточного припинення терапії у 4,4% пацієнтів; в інших хворих це небажане явище вдалося подолати шляхом застосування протидіарейної терапії, зменшення дози або призупинення лікування (див. розділ «Особливості застосування»).
Підвищення рівнів ферментів печінки
Підвищення рівнів ферментів печінки (див. розділ «Особливості застосування») було зафіксовано у 13,6% пацієнтів, які одержували терапію нінтеданібом. Підвищення рівнів ферментів печінки було оборотним і не пов’язаним із клінічно вираженим захворюванням печінки. Додаткова інформація стосовно особливих категорій пацієнтів, рекомендованих заходів та коригування доз у разі діареї та підвищення рівнів ферментів печінки наведена у розділах «Особливості застосування» та «Спосіб застосування та дози».
Повідомлення про побічні реакції
Повідомлення про побічні реакції після реєстрації лікарського препарату є важливими. Це дає змогу постійно вести моніторинг співвідношення користь/ризик лікарського засобу. Просимо медичних працівників повідомляти про будь-які можливі побічні реакції за допомогою національної системи звітності.
Термін придатності
3 роки.Умови зберігання
Зберігати при температурі не вище 25°C, в оригінальній упаковці для захисту від вологи.
Упаковка
По 10 капсул в блістері з алюмінієвої фольги з перфорацією, по 6 блістерів в картонній упаковці.
Категорія відпуску
За рецептом.
Виробник
Берінгер Інгельхайм Фарма ГмбХ і Ко. КГ/Boehringer Ingelheim Pharma GmbH & Co. KG.
Місцезнаходження
Бінгер Штрассе 173, 55216, Інгельхайм на Рейні, Німеччина
Binger Strasse 173, 55216, Ingelheim am Rhein, Germany.
Конец текста официальной инструкции
Дополнительная информация
Реклама препарата:
реклама препарата Офев на территории Украины запрещена.
Фармакотерапевтическая группа:
Антинеопластичні засоби. Інгібітори протеїнкінази.
Примечания
* - инструкция не переведена на русский язык. На странице предоставлена украиноязычная версия инструкции.
Количество просмотров: 224.